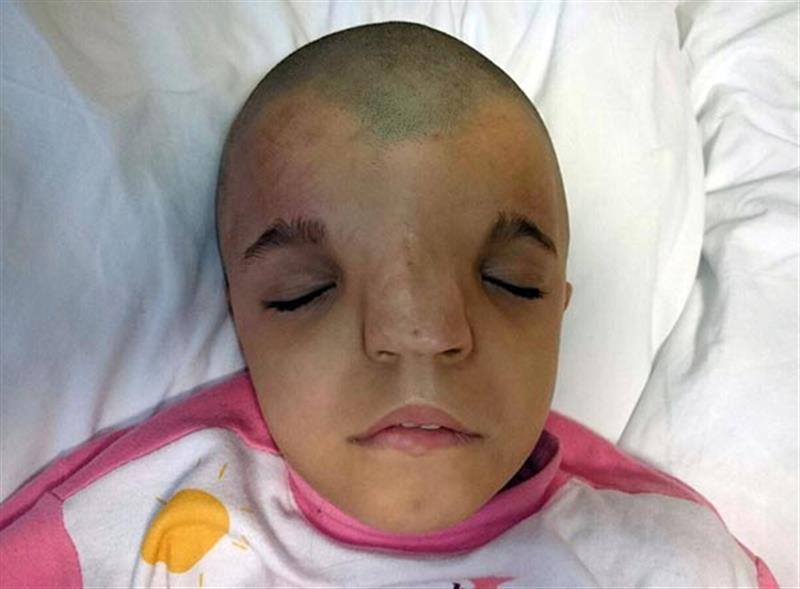
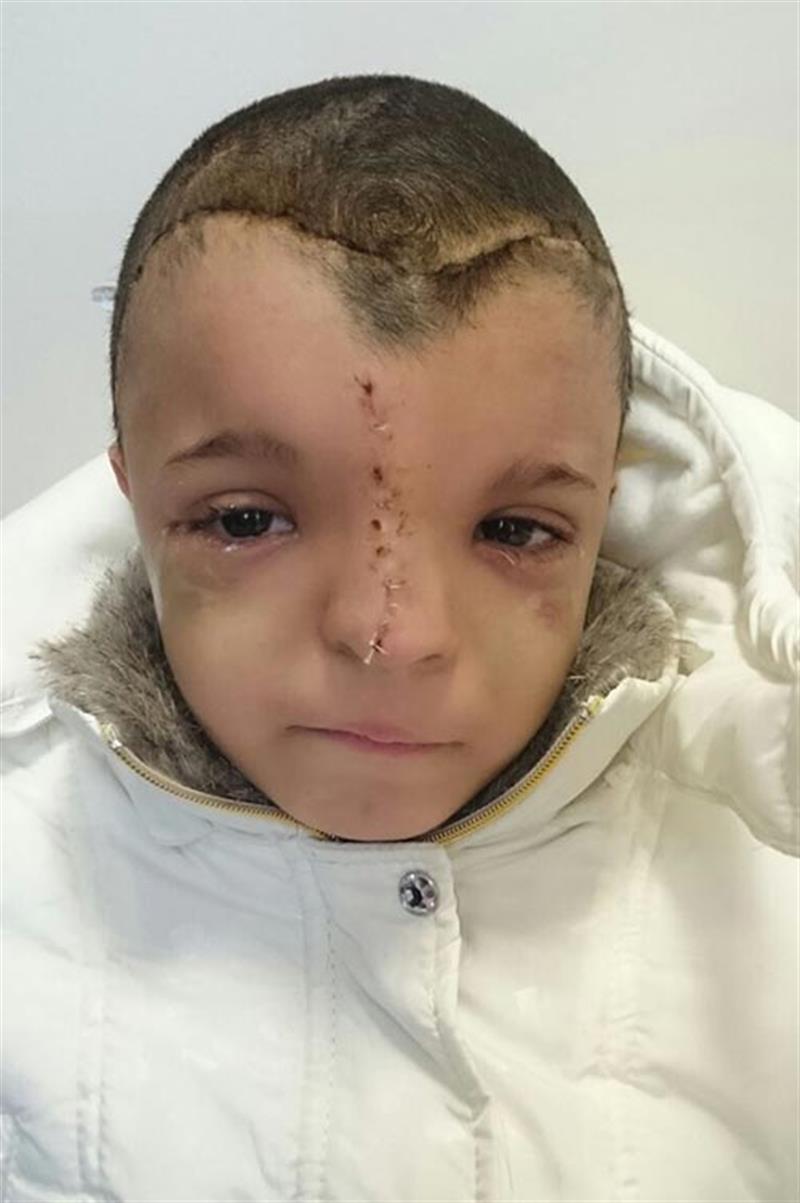
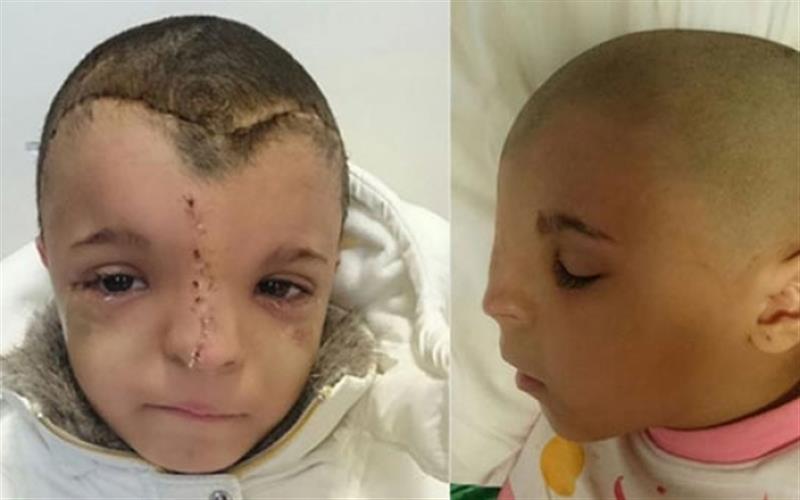
KRANİOSNESTOZ VE KRANİOFASİAL ANOMALİLER
(KAFA BURUN VE YÜZ KEMİKLERİ ANOMALİSİ)
Tanım : Kraniosnoztoz kafa tası kemiklerinin bir veya birden fazla eklem yerlerinin erken kapanmasıdır. Tarihçesi ilk defa 1791 yılında Sommering sonra 1852 de Virchow kafa tası kemiklerinin erken kapandığını tanımlamışlardır. Normalde kafa tası kemikleri arasındaki eklem yerleri öndeki ve arkadaki bıngıldak ( fontanel) denilen yerler bebeğin beyninin hacimsel olarak büyümesine ve hatta doğum esnasında doğumu kolaylaştıracak esnekliğe sahiptirler. Normal kafa tası kemikleri açık olarak doğan bir bebek ortalama 36 cm lik baş çevresi genişliği varken, bu çap yaklaşık 2 yaşında 49 cm ulaşmaktadır. Beyin dokusu hacim olarak değerlendirildiğinde doğumdaki beyin hacimi bebek 1 yaşına gelince 3 misline ulaşırken, iki yaşında ise doğum hacminin 4 katına ulaşmaktadır. Başka bir değişle beyin dokusu hacim olarak ilk 6 ayda erişkin beyin hacminin % 50 sine, 3 yaşında ise % 80 lere ulaşmaktadır. Böylece beyinin hacimsel gelişmesi sağlamak için kafa tası eklem yerleri açıklığı sayesinde oluşmaktadır. Kafa tası kemikleri eklem yerleri erken kapanması hem beyinsel hemde estetik problemlere neden olacaktır.
Kraniosneztozlar iki tipi vardır; 1 : Primer kraniosnostoz, 2 : Sekonder kraniosnoztoz.
PRİMER KRANİOSNESTOZLAR (nonsendromik ve sendromik) diye iki tipi vardır.
1: Non sendromik kraniosnoztoz, 2: Sendromik kraniosnostozlar.
Non sendromik kraniosnoztoz en sık görülen kraniosnoztoz tipidir. Bu tipi %80- 85 oranında görülür. En sık görülenleri;
1: Sagital sutur erken kapanmasına (skafo/ dolikosefali) başın ön arkaya doğru büyümesi olup tüm snostozların % 55 ini teşkil ederler. 1/ 5000 canlı doğumda görülmektedir.
2: Metopik sutur erken kapanması frontal kemikler arası eklemin kapanmasına (trigonosefali) adı verilir. Bu suturun kapanması 1/7000 veya 1/15000 doğumda görülür. Bu tip sutur erken kapanması başın üçgen şeklini almasına ve göz çukurlarının yüzeyeliğine neden olur.
3: Koroner suturlerin iki taraflı kapanması tüm kraniostozların % 20-30' unu teşkil ederlerken, bu tek taraflı olursa (anteriör plagiosefali) adı verilir. Anteriör plagiosefalide baş tek taraflı asimetrik olarak büyürken veya iki taraflı koroner sutur kapanırsa (oksisefali) denilen baş kule şeklinde yukarı doğru büyümesine neden olur. Daha nadiren % 2-3 oranında başın arkasındaki lamdoid suturler kapanabilir. Bu nadir görülen lamdoid sutur tek taraflı kapanırsa (posterior plagiosefali) adı verilir. Bu daha çok aynı pozisyonda yatırılan bebeklerin baş şekliyle karıştırırlır.
2: Sendromik kraniosnostozlar:
En sık sendromik kraniosneztoz çeşitleri;
1: Apert sendromu
2:Crouzon sendromu
3: Karpenter sendromu
4: Plefiffer sendromu ve bunlardan farklı olarak 100 üzerinde literatürde sendromik kraniosnestoz vakaları rapor edilmiştir. Bunlarda içerisinde Apert ve Crouzen sendromu en sık olanları olup 1/ 650000 veya bir milyon doğumda 15 olarak görülmektedir.
Kraniostozlarda kalıtım;
1: Otozomal dominant geçiş: bu erkek veya dişide ikişer adet olan bir gen mutasyonu sonucu ortaya çıkmaktadır. Otozomal dominant geçişli bir durumdan etekilenen bir bireyin her gebelikte normalin % 50 taşıyıcı olarak çocuğu etkilemesi beklenir. Kraniostozlu bir bebeğin anne ve babası bu pataloji açısından tetkik edilmelidir.
2: Otozomal resesif. az sayıda kraniostoz vakası böyle geçişlidir. Mutasyon oranı çok düşüktür. Her gebelikte % 25 rekkürens riski vardır.
3: X- bağlantılı
4: Kromozomal
5: Teratojenik (annenin valporat kullanımı)
6:bilinmeyen nedenler ( FGFR3, P2250R)
SEKONDER KRANİOSTOZLAR
Bu durum daha çok mikrosefali,şantlı hidrosefali li hastalar, hematolojik hastalıklar (rikets, D vitamin eksikliği), endokrin hastalıkaları (hipertiroidi, hiperkalsemi) sonrası görülürler.
Kraniosnestozlar Fizik ve Nörolojik değerlendirme;
Kafa tası tek bir sutur etkilendiğinde baş bu yönde büyüyemediğinde karşı tarafa büyüme olur en iyi örnek tek tarafalı koroner sutur kapandında (anterior plagiosefali) baş tek taraflı yukarı doğu büyür,veya iki taraflı koroner sutur etkilendiğinde baş yukarı doğru kule şeklinde büyür (oksisefali) görünümü, öndeki metopik sutur etkilendiğinde kafa tası öne üçgen şeklinde büyüme (trigonosefali), ortadaki sagital sutur etkilendiğinde baş ön ve arkaya doğru (dolikosefali), arkadaki lamdoid sutur etkilendiğinde başın arka kısmı baskılanmıştır ( posterior plagiosefali). Bu durum özellikle deformasyonal lamdoid snostoz özellikle başın dışarıdan gelen kuvvetlerin etkisiyle oluşan bir deformitedir. 300 canlı doğumda bir vakada görülür. En sık sebebi bebeklerin sırt üstü aynı pozisyonda yatırlılması sonrası görülür.Bu deformitenin gerçek lamdoid snostozdan ayırımı için klinik muayene yanında 3-boyulu bilgisyarlı tomografi yardımcıdır.
KRANİOSTOZLARDA KLİNİK BULGULAR
1: KİBAS: Kraniosnoztozların en ciddi klinik etkisi kafa içi basınç artımıdır. Bu durum tek sutur etkilendiğinde % 15, birden fazla sutur etkilendiğinde % 45 kadar çıkmaktadır. Özellikle bilaterel koroner ve sagital sutur etkilendğinde KİBA oranı % 92 kadar çıkmaktadır. KİBA başlıca; başağrısı, kusma, görme kaybı (papil ödemi, optik atrofi,şaşılık) görülebilir.
2:Mental değerlendirme: kraniostozlarda başın şekil bozukluğu yanında, mental ve ruhsal yetersizlikler görülür.Özellikle sendromik kraniostozlarda beyin gelişim etkilenmesi sonrası kognitif bozukluklar ve entellektüel gelişemsel yetersizlikler görülür. Sendromik kraniostozlarda IQ % 83 olarak bulunmuştur. Apert sendromunda mental bulgular daha ağırdır.
3: Hidrosefali:kraniostozlarda hidrosefali veya şanta bağımsız ventrikülomegali şeklinde iki şekilde görülür.Hidrosefali etkilenen sutur sayısına ve sendromik olup olmamasına göre değişir. Nonsendromik kraniostozlarda hidrosefali % 12 oranında görülürken bunların % 2-3 şant ihtiyacı göstermiştir. Sendromik kraniostozların ise % 44 oranında hidrosefali görülürken bunların % 6 'sı şant ihtiyaç göstermiştir. Hidrosefalinin nedeni olarak dar bir posterior fosa, küçük 4. ventrikül,venöz drenaj yetersizliğine bağlı BOS emilim bozukluğu teorileri ileri sürülmüştür.
4: Chiari sendromu: bu anomali hem sendromik hemde nonsendromik kraniostozlarda görülür. Tüm olguların içinde % 10 oranında Chiari anomalisi görülür. Bu oran en sık lamdoid suturde % 55 oranında görülürken, multi sutur etkilendiğinde oran % 57 iken, sagital suturde % 2, koroner sutur % 6 civarında görülür. Chiari malformasyonunda beyinden spinal kanala doğru BOS akımının kısmen engellenmesi, yanında küçük dar ve sığ bir posterior fossa kemiği senrası orta beyin KİBA sonrası beyincik aşağı doğru sarkmaktadır.Kraniosneostoz sıklıkla Chiari malformasyonuna eşlik eder.
5:Üst solunum yolu obstriksiyonu ( Obstriktif uyku apne sendromu )
özellikle sendromik kraniostozlarda doğumdan sonra ölüm nedeni üst solunum yolu tıkanması sonrası olur. Orta yüz hipoplazisi sonrası damak yukarı kaldırması sonrası hava yolunu daraltır böylece uyku apnesi olur.Uykuda CO2 retansionu sonrası KİBA ortaya çıkar. Bu durum sendomik kraniostozlarda % 40-68 oranında görülür.
KRANİOSTOZLARDA TEŞHİS
Teşhis öncelikle çocuk hekimlerinin çocuğun doğumundaki baş şekli, bıngıldakların (fontanel) açıklığı, baş çevresi ölçümleri, fontanel kabarıklığı gibi özel muayenesiyle başlar. Aylık çocuk gelişim takibinde baş şekli; başın gerek önde gerek arkaya asimetrik büyümesi varsa kraniostozdan şüphelendirmedir. Özelikle non sendromik ve tek sutur etkilenenlerde daha dikkatlice olunmalıdır. Sendromik kraniostozlarda çocuk doğar doğmaz bebekte; yüz, burun, göz, damak, dudak anomalisi varmı yokmu dikkalice kaydedilmelidir. Eğer sendromik kraniostoz ise daha seçici olarak doğru tanı tedavide çok yardımcıdır. Baş şekil bozukluğu ve beyinsel gelişimi, baş çevresi ve fontanel ölçümleri çocuk doğumdan sonraki resimleri çok dikkatli karşılaştılımalıdır. Burada temel soru kraniostoz varmı yokmu, varsa hangi tipi, kaç sutur etkilendiği, sedromik mi yoksa nonsendromik mi kraniostoz soruları dikkatlice cevaplandırılmalıdır. Teşhis yöntemleri ;
1: Baş çevre ölçümü,
2: Kafa tası suturleri palpasyonu ,
3: Kranial ultrasonografi,
4: 3 boyutlu Bilgi sayarlı Tomografi,
4: Beyin Magnetig Rezonans (MR) görüntüleme,
5 Kan testleri (hematolojik, endokrin testler),
TEDAVİ:
Bu anomalilerin tedavisi cerrahidir. Burada kapanan kafa tası kemikleri beyne bası yaparak beyin gelişmini engelemektedir. Bu basını önlenmesi ve giderilmesi amacı yanında, başın, yüzün, burun ve alın gibi şekil bozukluğunun düzeltilmesi kozmetik amaçtır.
Cerrahi tedavi zamanlaması;
1: Endoskopik sinektomiler ilk 3 ay içerisinde yapılmaktadır
2: Sagital sinostoz ( verteks rekonstruksionu 3 ay içerisinde yapılmaktadır
3: Frontal ve kalvarial sinostozlarda 4-12 aylık sürelerde,
4: Hipertelorizm 4-5 yaşlarında,
5: Lefort III ilerletme 5-7 yaşları arasında yapılmaktadır.
Sonuç olarak kraniostoz yenidoğan ve çocukluk dönemi nisbeten sık görülen bir hastalıktır. Bu anomalinin tanısı ve tedavisinde beyin cerrahı, plastik rekonstruksion cerrahı, anestezist, yeni doğan yoğun bakım hekim gibi tecrübeli hekim grublarının titiz ve sabırlı grub çalışmasıyla mükemmel sonuçlara ulaşılamaktadır.